Việt Nam là một trong những quốc gia có tỷ lệ mang gen/mắc bệnh Thalassemia cao trên thế giới. Ước tính mỗi năm có thêm khoảng 8.000 trẻ em sinh ra bị bệnh Thalassemia, trong đó có khoảng 2.000 trẻ mắc Thalassemia thể nặng và khoảng 800 trẻ không thể ra đời do phù thai, đây là gánh nặng về y tế, và xã hội hết sức nặng nề, đặc biệt là với những quốc gia đang phát triển như Việt Nam. Trung bình mỗi bệnh nhân Thalassemia thể nặng cần 470 đơn vị máu để sống đến 21 tuổi với chi phí điều trị lên đến hàng tỷ đồng. Hiện nay biện pháp hiệu quả nhất để tránh sinh con mắc Thalassemia thể nặng là chủ động sàng lọc bằng các xét nghiệm đơn giản, dễ thực hiện và chi phí thấp.
Các đối tượng cần sàng lọc Thalassemia bao gồm: Sàng lọc tiền hôn nhân, sàng lọc tiền mang thai, sàng lọc trước sinh đối với tất cả các cặp vợ chồng. Sàng lọc với những người có tiền sử gia đình mắc bệnh Thalassemia.
Quy trình sàng lọc Thalassemia được tiến hành qua từng bước, với xét nghiệm đầu tay là tổng phân tích tế bào máu ngoại vi. Những trường hợp nghi ngờ mang gen/mắc bệnh Thalassemia sẽ được chỉ định các xét nghiệm tiếp theo để đưa ra chẩn đoán xác định.
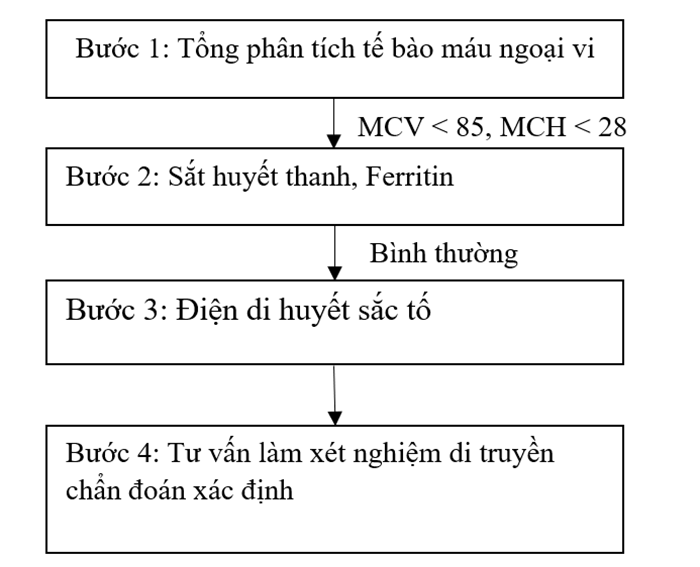
1. Tổng phân tích tế bào máu ngoại vi
Tổng phân tích tế bào máu ngoại vi là xét nghiệm được chỉ định đầu tay trong trường hợp sàng lọc/nghi ngờ người bệnh mắc Thalassemia. Sàng lọc được coi là dương tính khi MCV < 85fl và/hoặc MCH < 28pg. Ngoài MCV, MCH giảm, các biến đổi khác có thể xảy ra trên tổng phân tích tế bào máu ngoại vi gồm:
– Số lượng hồng cầu: Có thể tăng do tăng tạo máu, giảm hoặc bình thường.
– Hb: Có thể bình thường đối với người mang gen, và giảm với người bệnh Thalassemia.
– RDW: có thể bình thường/tăng.
Đây là xét nghiệm không đặc hiệu cho Thalassemia, nên không có giá trị chẩn đoán xác định. Cần thực hiện thêm các xét nghiệm khác để chẩn đoán phân biệt và chẩn đoán xác định. Cần lưu ý, MCV, MCH trong giới hạn bình thường không giúp loại trừ tình trạng mang gen Thalassemia. Một số trường hợp MCV, MCH bình thường, vẫn có thể mang gen thalassemia do sai số của xét nghiệm công thức máu, bệnh lý kèm theo,…
2. Xét nghiệm sinh hóa máu: Sắt huyết thanh, Ferritin
Sắt huyết thanh và Ferritin có giá trị phân biệt Thalassemia với các bệnh thiếu máu hồng cầu nhỏ, nhược sắc khác. Đối với người bệnh/người mang gen Thalassemia, sắt huyết thanh và Ferritin thường nằm trong giới hạn bình thường. Một số trường hợp Ferritin có thể tăng do quá tải sắt.
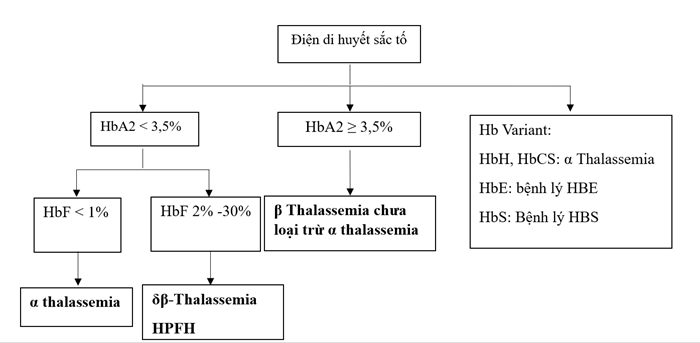
3. Điện di huyết sắc tố
Điện di huyết sắc tố có giá trị định hướng thể bệnh nhằm đưa ra các xét nghiệm di truyền phù hợp.
Các loại huyết sắc tố ở người bình thường gồm:
– Huyết sắc tố A (HbA, α2β2) chiếm tỷ lệ cao nhất, gặp ở trẻ em và người trưởng thành.
– Huyết sắc tố F (HbF, α2γ2) (huyết sắc tố thai nhi) thường được tìm thấy trong các bào thai đang phát triển. Ở trẻ sơ sinh, HbF vẫn chiếm tỷ lệ lớn. Lượng HbF nhanh chóng giảm xuống khi trẻ được 1 tuổi, ở người trưởng thành, HbF <1%.
– Huyết sắc tố A2 (HbA2, α2β2) là loại huyết sắc tố bình thường được tìm thấy với số lượng nhỏ ở người trưởng thành.
Kết quả điện di huyết sắc tố bình thường theo tuổi
| Lứa tuổi | HbA (%) | HbA2 (%) | HbF (%) |
| Sơ sinh | 20 – 40 | 0,03 – 0,6 | 60 – 80 |
| 2 tháng | 40 – 70 | 0,9 – 1,6 | 30 – 60 |
| 4 tháng | 80 – 90 | 1,8 – 2,9 | 10 – 20 |
| 6 tháng tuổi | 93 – 97 | 2 – 3 | 1 – 5 |
| 1 tuổi | 97 | 2 – 3 | 0,4 – 2 |
| >5 tuổi | 97 | 2 – 3 | 0,4 – 2 |
| Người trưởng thành | 96 – 98 | 0,5 – 3,5 | < 1 |
Thay đổi điện di huyết sắc tố trong bệnh Thalassemia:
– Alpha Thalassemia thể ẩn không có thay đổi trên kết quả điện di huyết sắc tố. Thể HbH có HbA bình thường hoặc giảm, xuất hiện HbH với tỷ lệ 0,8% – 40%, có thể xuất hiện các huyết sắc tố khác như HbCS, HbQS, ….
– Khác với α thalassemia, β Thalassemia có thay đổi rõ rệt trên kết quả điện di huyết sắc tố, với HbA giảm, HbA2 tăng, HbF tăng. Mức độ thay đổi khác nhau giữa các thể lâm sàng:
+ β Thalassemia thể nhẹ: HbA trong giới hạn bình thường, HbA2 ≥ 3,5%, HbF bình thường/tăng nhẹ.
+ β Thalassemia thể trung gian: HbA giảm, HbA2 ≥ 3,5%, HbF tăng nhiều (10% -50%).
+ β Thalassemia thể nặng: HbF chiếm ưu thế, có thể lên đến 100%.
4. Xét nghiệm di truyền
Xét nghiệm di truyền có giá trị chẩn đoán xác định Thalassemia, xác định loại đột biến gây bệnh, từ đó có thể đưa ra tư vấn phù hợp nhằm tránh sinh con mắc Thalassemia thể nặng. Hiện nay, có rất nhiều xét nghiệm di truyền có thể ứng dụng trong chẩn đoán Thalassemia, phạm vi khảo sát của các xét nghiệm khác nhau, mỗi kỹ thuật đều có ưu điểm và giới hạn nhất định. Dựa vào xét nghiệm tổng phân tích tế bào máu ngoại vi và điện di huyết sắc tố, bác sĩ sẽ chỉ định xét nghiệm di truyền phù hợp với từng bệnh nhân nhằm tiết kiệm thời gian và chi phí xét nghiệm.

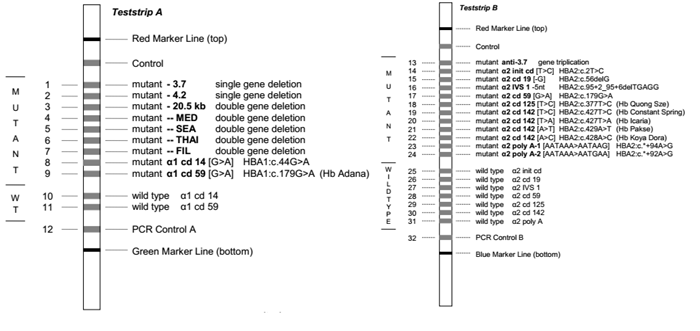
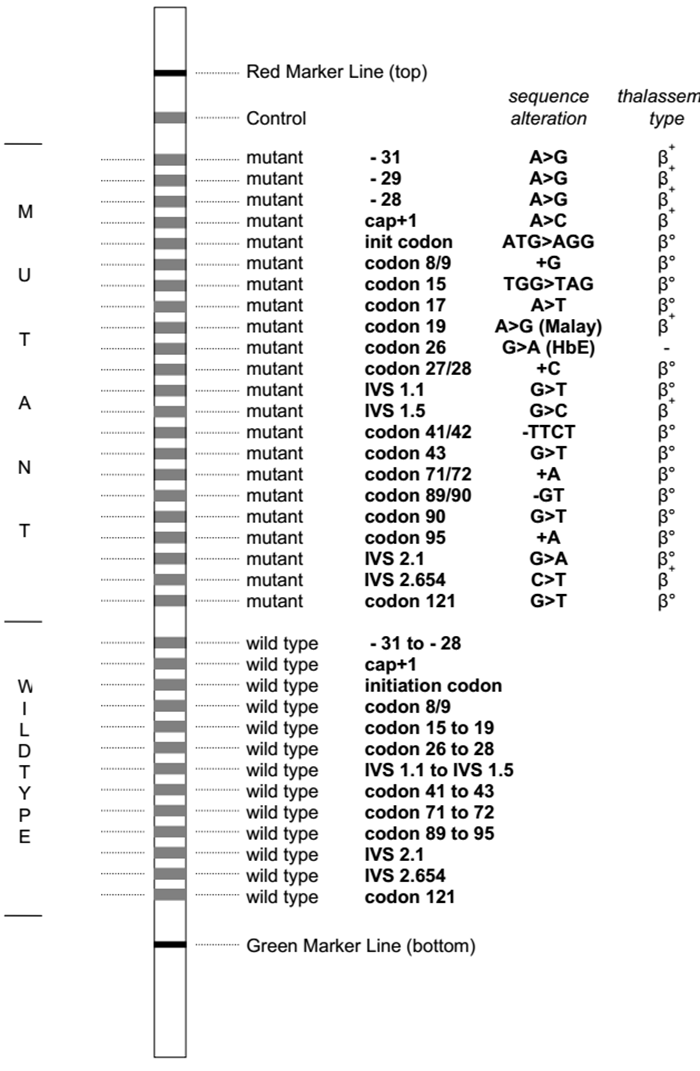
4.1. Xét nghiệm lai phân tử strip assay
Xét nghiệm lai phân tử dựa trên nguyên lý sử dụng các đầu dò đặc hiệu alen (allele specific oligonucleotide – ASO probes) bình thường và đột biến được gắn cố định trên các dải nitrocellulose (nitrocellulose strips) có màng nylon, lai với các mẫu DNA cần phân tích nhằm khảo sát nhiều đột biến cùng lúc.
Sản phẩm DNA sau khuếch đại bằng PCR được đánh dấu bằng biotin, sau đó được biến tính và lai với các đầu dò cố định. Sau quá trình rửa nghiêm ngặt, sản phẩm hiển thị dưới dạng các vạch trên thanh lai, có thể quan sát và đánh giá bằng mắt thường.
Ưu điểm của kỹ thuật này là có thể khảo sát cùng lúc các đột biến điểm và đột biến mất đoạn hay gặp, gồm 21 đột biến gen α globin (7 đột biến mất đoạn, 1 đột biến lặp đoạn và 13 đột biến điểm) và 22 đột biến điểm trên gen β globin hay gặp nhất (hình 2), chiếm 90% các đột biến gây bệnh Thalassemia hay gặp ở khu vực Đông Nam Á với độ nhạy, độ đặc hiệu cao. Tuy nhiên, nhược điểm của kỹ thuật này là giá thành xét nghiệm tương đối cao và không có khả năng phát hiện các đột biến hiếm gặp.
Do đó, hiện nay strip assay được chỉ định như xét nghiệm di truyền đầu tay để phát hiện các đột biến gây α Thalassemia hoặc β Thalassemia hay gặp. Trong trường hợp strip assay không phát hiện đột biến, các kỹ thuật khác có thể được chỉ định để mở rộng phạm vi khảo sát.
4.2. Gap PCR
Kỹ thuật Gap PCR sử dụng hai đoạn mồi bổ sung với mạch xuôi và mạch ngược ở hai vùng biên đoạn ADN bị đứt. Đối với đột biến mất đoạn lớn, mồi thiết kế ở hai vùng biên chỉ khuếch đại được alen đột biến mất đoạn, bởi vì khoảng cách giữa hai mồi quá lớn không thể khuếch đại alen bình thường. Trường hợp alen không đột biến có thể phát hiện bằng cách thiết kế một mồi bổ sung với trình tự trong vùng mất đoạn, một mồi bổ sung với trình tự ADN vùng biên.
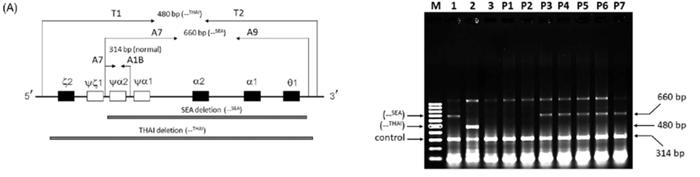
Hiện nay, tại Việt Nam, bộ kit xét nghiệm Gap PCR cho phép phát hiện 5 đột biến mất đoạn α Thalassemia hay gặp, bao gồm: SEA, -3.7, -4.2, FIL, THAI.
Ưu điểm của GAP-PCR là kỹ thuật đơn giản, thời gian xét nghiệm nhanh, và chi phí thấp. Tuy nhiên, GAP-PCR chỉ cho phép khảo sát một số đột biến mất đoạn hay gặp và không có khả năng phát hiện đột biến điểm, các đột biến mất đoạn hiếm gặp và đột biến mất đoạn mới do yêu cầu phải biết chính xác điểm đứt để thiết kế mồi.
Với những ưu nhược điểm trên, trong thực hành lâm sàng, Gap PCR thường được chỉ định khi kết quả điện di huyết sắc tố nghi ngờ α Thalassemia, nhằm phát hiện các đột biến mất đoạn phổ biến.
4.3. MLPA
Kỹ thuật MLPA sử dụng các đoạn dò (probe) có khả năng lai với phân tử DNA đích đặc hiệu. Mỗi đoạn dò (probe) của phương pháp MLPA bao gồm 2 phần: đoạn oligonucleotide phái bên trái (left probe oligonucleotide – LPO) và bên phải (right probe oligonucleotide – RPO). Trên các đoạn LPO và RPO có chứa trình tự mồi cho phản ứng PCR và trình tự lai DNA. Một đoạn trình tự sẽ được thêm vào phía bên đoạn RPO để khiến cho mỗi đoạn dò có một kích thước đặc biệt. Sản phẩm lai sau đó được phát hiện thông qua kỹ thuật điện di mao quản. Hiện nay, có 2 bộ promix chứa các probe dành riêng cho alpha Thalassemia và β Thalassemia, gồm:
– Probemix P140 HBA gồm 45 probe MLPA có chiều dài khác nhau, thay đổi từ 130 – 481 nucleotid, giúp phát hiện hầu hết các đột biến mất đoạn, một số đột biến lặp đoạn và đột biến điểm hay gặp nhất (HbCS) trên gen α Globin.
– Probemix P102 HBB gồm 49 probe MLPA có kích thước khác nhau, thay đổi từ 130 – 502 nucleotid, trong đó có 40 probe cho vùng gen HBB và các vùng lân cận và 9 probe cho các vùng tham chiếu thuộc các NST khác, cho phép phát hiện hầu hết các đột biến mất đoạn trên gen cụm gen beta gồm HBG1, HBG2, HBB và HBD liên quan đến nhóm bệnh beta-thalassaemia.
Ưu điểm của kỹ thuật MLPA là cho phép phát hiện cả các đột biến mất đoạn và lặp đoạn đã biết và chưa biết, hay gặp và ít gặp. Nhược điểm của kỹ thuật MLPA là không có khả năng phát hiện đột biến điểm (trừ HbCS) và giá thành xét nghiệm cao.
Do các đặc điểm trên, trong thực hành lâm sàng MLPA chủ yếu được chỉ định trong trường hợp nghi ngờ α Thalassemia do mất đoạn, tuy nhiên không phát hiện đột biến mất đoạn hay gặp trên Gap PCR, nhằm mở rộng phạm vi khảo sát hoặc β Thalassemia/ δβ Thalassemia nghi do mất đoạn trên cụm gen β Globin.
Trên đây là những thông tin cần biết về xét nghiệm xác định đột biến gen tan máu bẩm sinh Thalassemia. Nếu có thắc mắc cần bác sĩ Phương Hoa tư vấn, giải đáp, vui lòng liên hệ.

Bác sĩ Phương Hoa
Chuyên gia di truyền y học
Liên hệ với bác sĩ





Website này được xây dựng với mong muốn lan tỏa những thông tin hữu ích trong lĩnh vực di truyền học, trở thành kênh kết nối chuyên môn giữa các bác sĩ, chuyên gia y tế nhằm mang lại sự hỗ trợ tốt nhất cho người bệnh.