Thalassemia hay bệnh thiếu máu tan máu bẩm sinh là một trong những bệnh di truyền phổ biến ở Việt Nam. Nếu không được phát hiện sớm và có cách điều trị phù hợp, bệnh sẽ gây ảnh hưởng nghiêm trọng đến sức khỏe, chất lượng cuộc sống. Nhiều trường hợp ở thể nặng thậm chí cần chăm sóc y tế cả đời, kéo theo gánh nặng cho cả gia đình và xã hội.
1. Thalassemia là gì?
Thalassemia là bệnh thiếu máu tan máu bẩm sinh di truyền do thiếu hụt tổng hợp một hoặc nhiều chuỗi globin.
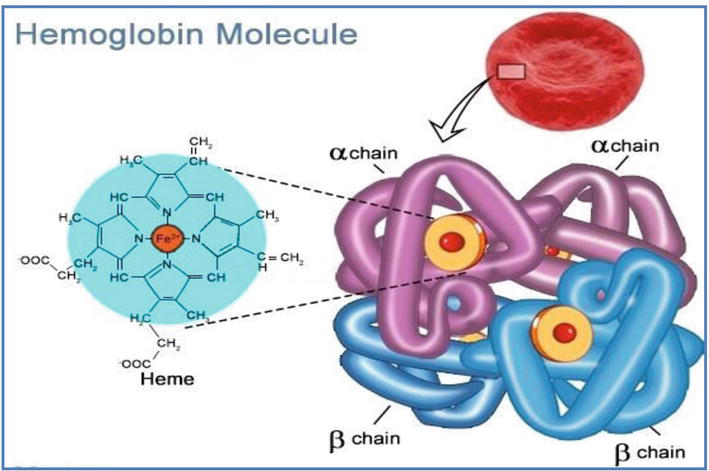
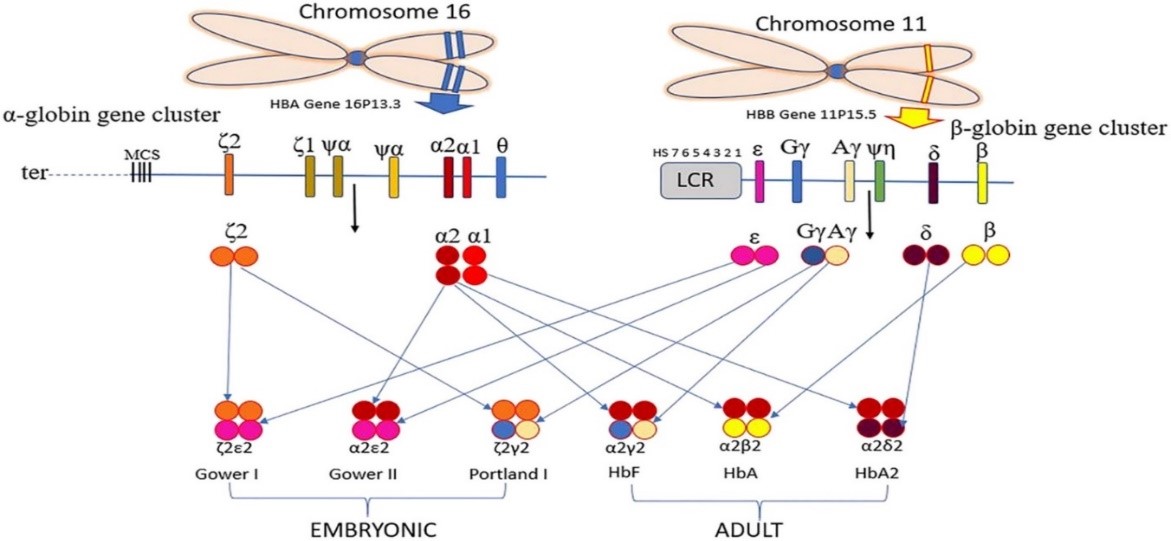
Phân tử Hemoglobin bình thường được cấu tạo bởi nhân Hem và 4 chuỗi Globin, gồm 2 chuỗi α và 2 chuỗi khác không phải α, ví dụ β Globin. Trong giai đoạn bào thai, Hemoglobin trong máu thai là HbF (cấu tạo bởi 2 chuỗi α và 2 chuỗi γ), sau sinh, HbF dần bị thay thế bởi HbA (2 chuỗi α và 2 chuỗi β) và HbA2 (2 chuỗi α và 2 chuỗi δ). Tỷ lệ huyết sắc tố ở người lớn bình thường: HbA1: 97-98%, HbA2: 2-3%, HbF: dưới 1%.
Dựa vào loại globin bị thiếu hụt tổng hợp, Thalassemia được chia thành 2 nhóm: α-Thalassemia: do thiếu hụt tổng hợp chuỗi α Globin và β-Thalassemia: do thiếu hụt tổng hợp chuỗi β Globin.
Thalassemia là một trong những rối loạn di truyền phổ biến nhất thế giới. Hiện nay, ước tính khoảng 7% dân số thế giới mang gen Thalassemia và 1,1% các cặp vợ chồng có nguy cơ sinh con mang gen/bị bệnh Thalassemia. Bệnh phân bố khắp các khu vực trên thế giới, phổ biến ở vùng Địa Trung Hải, Trung Đông, châu Á – Thái Bình Dương, trong đó Việt Nam là một trong những nước có tỷ lệ mắc bệnh và mang gen bệnh cao. Tại Việt Nam, theo nghiên cứu của Viện Huyết học truyền máu Trung ương năm 2017, tỷ lệ người mang gen/mắc bệnh Thalassemia trên quần thể dân số chung là 13,8%, phân bố khác nhau giữa các dân tộc, thấp hơn ở người Kinh với 2% – 4%, và đặc biệt cao ở nhóm dân tộc thiểu số: tỷ lệ ở người Mường là 22%, và trên 40% ở dân tộc Tày, Thái, ….
2. Nguyên nhân
Thalassemia là bệnh di truyền lặn trên nhiễm sắc thể thường, gây ra do đột biến gen mã hóa chuỗi globin, gồm gen α-globin (gây bệnh α-Thalassemia) và gen β-globin (gây bệnh β-Thalassemia).
Mỗi người có 4 bản sao của gen α globin (αα/αα), nằm trên cụm gen α của NST 16 (cụm gen này gồm hai gen α (α1 – HBA1, α2 – HBA2), một gen phôi (δ2 – HBE), 3 giả gen (ψδ1, ψα2, ψα1), và 1 gen chưa xác định được chức năng (θ1). Các gen này sắp xếp theo trình tự từ đầu 5’ đến đầu 3’ như sau: 5’- δ2 – ψδ1 – ψα2 – ψα1 – α2 – α1 – θ1 – 3’).
Hiện nay, khoảng >70 đột biến gen α globin đã được xác định, trong đó 85% là đột biến mất đoạn, phần còn lại là đột biến điểm (đột biến gây thay đổi một/một vài nucleotide). Tại Việt Nam, theo thống kê của viện huyết học truyền máu Trung Ương, 5 đột biến α Thalassemia hay gặp nhất gồm: SEA, -3.7, -4.2, HbCS, HbQS.
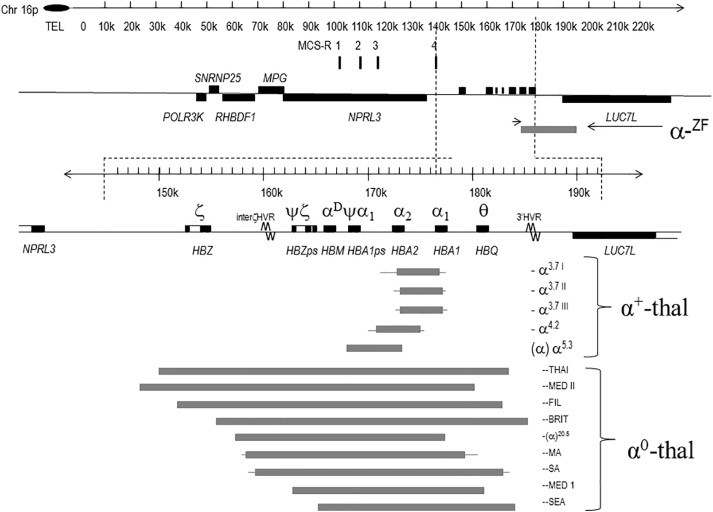
Cụm gen β globin nằm trên nhánh ngắn NST 11, gồm 5 gen sắp xếp theo trình tự 5′ ɛ, Gγ, Aγ, δ, β 3’. β-Thalassemia gây ra do đột biến trên gen β Globin, gây giảm hoặc mất khả năng tổng hợp chuỗi β Globin.
Khoảng 300 đột biến gen β-globin đã được xác định, phần lớn là đột biến điểm, các đột biến mất đoạn rất hiếm gặp. Dựa vào mức độ giảm tổng hợp chuỗi β-globin, đột biến gen β-globin được chia thành 3 nhóm: β0: mất hoàn toàn khả năng tổng hợp chuỗi β-globin, β+: giảm mạnh khả năng tổng hợp chuỗi β-globin (còn khoảng 10%), β++: khả năng tổng hợp chuỗi β-globin giảm không đáng kể.
Bảng 1. Các đột biến gen α-globin và β-globin hay gặp ở Việt Nam
| Gen α-globin | Phân loại | Gen β-globin | Phân loại |
| α3.7 (mất đoạn 3.7 kb trên gen α globin) | α+ | Codon 26 (HBE G>A) | β++ |
| α4.2 (mất đoạn 4.2 kb trên gen α globin) | α+ | Cd41/42 (-TCTT) | β0 |
| SEA (mất đoạn 20.5 kb gồm toàn bộ gen HBA1 và gen HBA2) | α0 | Cd17 (A>T) | β0 |
| HbCS (đột biến điểm gây thay đổi nucleotid T>C trên codon kết thúc của gen HBA2 dẫn tới chuỗi α globin tăng thêm 31 axit amin) | α+ | Cd95 (+A) | β0 |
| HbQS (đột biến điểm T>C trên codon 125 của gen HBA2 gây thay đổi axit amin Leucin thành Prolin) | α+ | Cd8/9 (+G) | β0 |
| Cd71/72 (+ A) | β0 | ||
| IVS1-1 (G>T) | β0 | ||
| IVS1-5 (G>C) | β0 | ||
| -28 (A>G) | β+ |
Ghi chú: α 0: đột biến mất 2 gen α globin trên một NST, α+: đột biến trên 1 gen α globin.
3. Triệu chứng
Biểu hiện của Thalassemia thay đổi từ nhẹ đến nặng tùy thuộc đột biến gen gây bệnh và mức độ thiếu hụt chuỗi Globin.
-
α Thalassemia:
Dựa vào số lượng gen α globin bị đột biến, α Thalassemia được chia thành 4 thể lâm sàng:
α Thalassemia thể ẩn: (α+/αα)
Đây là thể nhẹ nhất của α Thalassemia, gây ra do đột biến 1 gen α globin. Người mang gen thể ẩn thường không có biểu hiện lâm sàng, chủ yếu được phát hiện tình cờ khi xét nghiệm tổng phân tích tế bào máu ngoại vi, với MCV giảm nhẹ (<85 fl) và MCH giảm nhẹ (<28 pg/l). Thể này không có thay đổi trên kết quả điện di huyết sắc tố.
α Thalassemia thể nhẹ: (α0/αα, α+/ α+)
α Thalassemia thể nhẹ gây ra do đột biến mất 2 gen α globin, có thể trên một NST (α0/αα), hoặc trên hai NST tương đồng (α+/ α+). Bệnh nhân mắc α-thalassemia thể nhẹ thường có biểu hiện thiếu máu nhẹ: da xanh, mệt mỏi. Trên xét nghiệm tổng phân tích tế bào máu ngoại vi, bệnh nhân có biểu hiện hồng cầu nhỏ, nhược sắc, MCV giảm, MCH giảm. Thể này không có thay đổi trên kết quả điện di huyết sắc tố.
HbH (α0/ α+)
HbH gây ra do đột biến mất 3 gen α globin, gồm 2 thể: HbH thể mất đoạn (–/-α) gây ra do bệnh nhân mang 2 đột biến mất đoạn và HbH thể không mất đoạn gây ra do bệnh nhân mang một đột biến mất đoạn và một đột biến điểm (–/αT), các nghiên cứu cho thấy thể không mất đoạn thường có biểu hiện nặng hơn so với thể mất đoạn.
Triệu chứng lâm sàng thường gặp nhất ở bệnh nhân HbH là tình trạng thiếu máu, mức độ thiếu máu thay đổi từ nhẹ đến nặng. Bệnh nhân thường có lách to, vàng da ở các mức độ khác nhau. Trẻ mắc HbH có thể có biểu hiện chậm lớn. Ngoài ra còn có thể có các dấu hiệu do biến chứng khác của bệnh như: nhiễm trùng, các vết loét ở chi dưới, và có thể có các cơn tan máu cấp sau khi điều trị thuốc hoặc sau các đợt nhiễm trùng nặng. Bệnh nhân lớn tuổi hơn có thể có biểu hiện ứ sắt ở nhiều mức độ khác nhau.
Xét nghiệm tổng phân tích tế bào máu ngoại vi cho thấy tình trạng thiếu máu hồng cầu nhỏ nhược sắc từ trung bình đến nặng. Kết quả điện di huyết sắc tố lượng HbH thay đổi từ 0,8-40%, đôi khi còn có kèm theo Hb Bart’s ở một vài trường hợp.
Hb Bart (α0/ α0)
Hội chứng phù thai do Hb Bart’s là thể bệnh nặng nhất của α-thalassemia, gây ra do mất hoàn toàn 4 gen α globin, dẫn tới tình trạng mất hoàn toàn khả năng sản xuất chuỗi α globin. Ở thai mắc Hb Bart’s, thành phần Hb trong hồng cầu chủ yếu là Hb không có chức năng γ4 và β4. Thai mắc Hb Bart’s có biểu hiện phù thai, suy tim và thiếu máu kéo dài từ giai đoạn thai trong tử cung. Ngoài ra còn biểu hiện gan lách to, não chậm phát triển, hệ xương và hệ tim mạch phát triển bất thường. Trẻ mắc Hb Bart’s hầu hết thường tử vong ngay trong giai đoạn thai (23-38 tuần) hoặc ngay sau khi sinh. Đến nay, chỉ ghi nhận một vài trường hợp trẻ Hb Bart’s sinh ra sống nhờ được điều trị tích cực và truyền máu ngay trong giai đoạn sơ sinh.
-
β Thalassemia
β Thalassemia thể nhẹ (β0/β, β+/β)
Phần lớn bệnh nhân β Thalassemia thể nhẹ thường không có biểu hiện lâm sàng, một số trường hợp có thể có biểu hiện thiếu máu nhẹ. Trên tổng phân tích tế bào máu ngoại vi, bệnh nhân có biểu hiện thiếu máu hồng cầu nhỏ nhược sắc, với Hgb, MCV, MCH giảm nhẹ. Điện di huyết sắc tố có HbA giảm nhẹ/bình thường, HbA2 tăng, HbF bình thường/tăng nhẹ.
β Thalassemia thể trung gian (β0/β+, β+/β+)
β Thalassemia thể trung gian khởi phát muộn hơn so với thể nặng. Phổ kiểu hình của thể này tương đối thay đổi, từ thiếu máu nhẹ đến thiếu máu mức độ trung bình, vàng da, gan lách to, biến dạng xương, chậm phát triển thể chất. Các biến chứng do ứ đọng sắt như suy tim, xơ gan có thể xuất hiện nhưng muộn hơn thể nặng. Thể này thường không yêu cầu truyền máu kéo dài, thông thường bệnh nhân chỉ cần truyền máu khi có các vấn đề kèm theo như nhiễm trùng, dùng thuốc…
Kết quả tổng phân tích tế bào máu ngoại vi cho thấy tình trạng thiếu máu hồng cầu nhỏ, nhược sắc, Hgb, MCV, MCH giảm. Ferritin có thể tăng do ứ đọng sắt. Điện di huyết sắc tố có HbA giảm, HbA2 tăng, HbF tăng (10%-50%).
β Thalassemia thể nặng (β0/β0, β0/β+)
β Thalassemia thể nặng thường khởi phát sớm, từ 6 – 24 tháng tuổi. Trẻ mắc thể này thường có biểu hiện thiếu máu nặng, phụ thuộc truyền máu, vàng da, gan lách to. Nếu không được truyền máu định kỳ, trẻ thường có biến chứng chậm lớn, loét chân, phát triển các khối u ngoài tủy, biến dạng xương, bộ mặt thalassemia, cuối cùng là tử vong do suy tim. Truyền máu kéo dài kèm theo tình trạng tăng hấp thu sắt ở bệnh nhân Thalassemia có thể dẫn tới ứ đọng sắt ở các cơ quan nội tạng, gây tình trạng quá tải sắt với biểu hiện suy tim do ứ đọng sắt ở tim, viêm gan, xơ gan do ứ đọng sắt ở gan.
Kết quả tổng phân tích tế bào máu ngoại vi cho thấy tình trạng thiếu máu hồng cầu nhỏ, nhược sắc, Hb, MCV, MCH giảm nhiều. Ferritin có thể tăng do ứ đọng sắt. Điện di huyết sắc tố có HbA giảm rất thấp, HbF tăng cao (có thể lên đến 100%).
4. Sàng lọc và chẩn đoán bệnh Thalassemia
Ø Tổng phân tích tế bào máu ngoại vi
Tổng phân tích tế bào máu ngoại vi được coi là xét nghiệm đầu tay để phát hiện người bệnh và sàng lọc người mang gen bệnh thalassemia. Trên kết quả tổng phân tích tế bào máu ngoại vi, bệnh nhân/người mang gen Thalassemia thường có biểu hiện thiếu máu hồng cầu nhỏ, nhược sắc. Mức độ giảm Hb, MCV, MCH tuỳ thuộc vào loại từng thể bệnh.
Ø Sắt huyết thanh, Ferritin
Sắt huyết thanh và Ferritin có giá trị phân biệt Thalassemia với các bệnh thiếu máu hồng cầu nhỏ, nhược sắc khác. Đối với người bệnh/người mang gen Thalassemia, sắt huyết thanh và Ferritin thường nằm trong giới hạn bình thường. Một số trường hợp Ferritin có thể tăng do quá tải sắt.
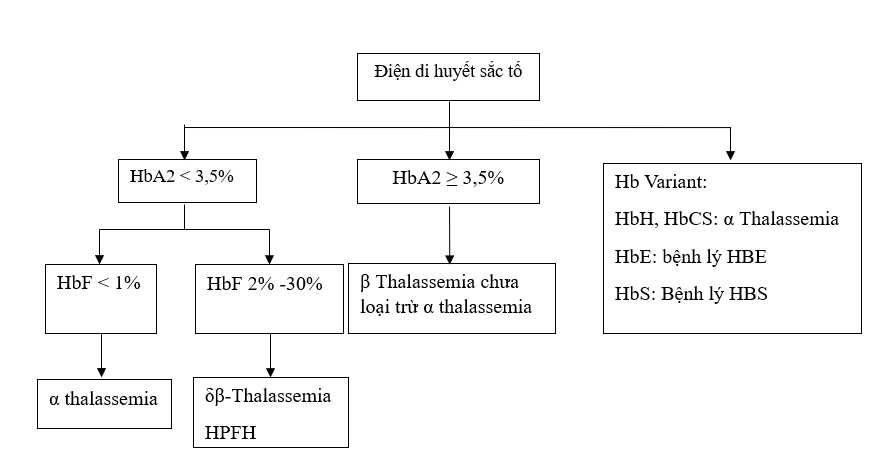
Ø Điện di huyết sắc tố
Dựa vào kết quả điện di huyết sắc tố, bác sĩ có thể xác định bệnh nhân thuộc nhóm Thalassemia nào để chỉ định các xét nghiệm di truyền phù hợp.
Ø Xét nghiệm di truyền
Xét nghiệm di truyền có giá trị chẩn đoán xác định, chẩn đoán thể bệnh và tiên lượng bệnh nhân. Hiện nay, có nhiều kỹ thuật khác nhau được ứng dụng trong chẩn đoán Thalassemia.
- Xét nghiệm lai phân tử ngược ASO (Stripassay) cho phép phát hiện 21 đột biến αThalassemia và 22 đột biến β Thalassemia hay gặp
- Xét nghiệm xác định đột biến mất đoạn: Gap PCR, MLPA.
- Xét nghiệm xác định đột biến điểm: ARMS PCR, Sanger Sequencing, NGS.
5. Điều trị
Điều trị thiếu máu
- Chỉ định truyền máu được đặt ra khi bệnh nhân có Hb < 7g/dL sau 2 lần kiểm tra, cách nhau > 2 tuần (sau khi loại trừ nguyên nhân khác như thiếu sắt và nhiễm trùng kèm theo) và/hoặc có biểu hiện: Chậm phát triển, biến dạng xương, gan lách to, sạm da.
- Chế phẩm: hồng cầu lắng phù hợp nhóm máu ABO và Rh.
- Mục tiêu: duy trì Hb khoảng 90 g/L
Thải sắt
- Chỉ định: Thải sắt được chỉ định khi ferritin máu > 1000ng/ml, hoặc sau truyền máu 10-20 lần.
- Phương pháp thải sắt: Thải sắt bằng thuốc truyền dưới da Desferrioxamine. Thời gian truyền từ 8 – 12 giờ/ đêm trong 5- 6 đêm/tuần.
Cắt lách
Cắt lách cần được cân nhắc trước khi tiến hành ở bệnh nhân Thalassemia do bệnh nhân có khả năng tăng nguy cơ huyết khối tĩnh mạch, tăng áp phổi, và nguy cơ nhiễm trùng nặng sau cắt lách. Cắt lách chỉ nên chỉ định trong trường hợp truyền máu không hiệu quả, cường lách, lách to (lách to độ IV).
Sau cắt lách bệnh nhân cần được tiêm phòng và điều trị dự phòng bằng kháng sinh liên tục đến năm 16 tuổi.
Liệu pháp ghép tế bào gốc tạo máu
Ghép tế bào gốc tạo máu được xem là phương pháp duy nhất có khả năng điều trị khỏi Thalassemia hiện nay. Chỉ định ghép tế bào gốc được đặt ra trong trường hợp bệnh nhân Thalassemia thể nặng phụ thuộc truyền máu, bệnh nhân trẻ tuổi (tối ưu nhất là dưới 14 tuổi), khi chưa có quá tải sắt mức độ nặng và có người cho tế bào gốc phù hợp HLA. Tỷ lệ thành công trong trường hợp này lên đến 90% và tỷ lệ tử vong là 4%.
Nguồn tế bào gốc có thể sử dụng gồm sử dụng nguồn tế bào gốc từ máu dây rốn, máu ngoại vi hoặc tủy xương của người cho hòa hợp HLA. Trong đó ghép tế bào gốc từ tủy xương và máu cuống có tiên lượng tốt hơn máu ngoại vi do nguy cơ gây ra bệnh mảnh ghép chống ký chủ cấp tính (GVHD cấp) thấp hơn.
Tỷ lệ sống sót sau ghép tế bào gốc là 90% và tỷ lệ khỏi hoàn toàn Thalassemia là 70-80%. Tỷ lệ tử vong sau ghép tế bào gốc tạo máu lên đến 10%. Nguyên nhân tử vong có thể do biến chứng trong quá trình ghép (suy tủy, suy gan, suy thận do điều trị hóa chất) hoặc sau quá trình ghép (bệnh ghép chống chủ (Graft vs Host disease), trong đó các tế bào của mảnh ghép tấn công các cơ quan trong cơ thể bệnh nhân như da, gan, niêm mạc đường tiêu hóa, phổi).
6. Phòng bệnh
Sàng lọc tiền hôn nhân, sàng lọc tiền mang thai và sàng lọc trước sinh bằng tổng phân tích tế bào máu ngoại vi, sắt huyết thanh và Ferritin cho các cặp vợ chồng nhằm phát hiện người mang gen bệnh là phương pháp có giá trị nhất trong phòng ngừa Thalassemia.
Các trường hợp Thiếu máu hồng cầu nhỏ, nhược sắc có sắt huyết thanh và Ferritin bình thường cần được làm xét nghiệm điện di huyết sắc tố và xét nghiệm di truyền để xác định đột biến.
Khuyến cáo tư vấn và chẩn đoán trước sinh/ chẩn đoán tiền làm tổ cho các cặp vợ chồng có nguy cơ sinh con mắc Thalassemia thể nặng (Hb Bart, HbH, β Thalassemia thể nặng).
Trên đây là những thông tin tổng hợp về Thalassemia – bệnh thiếu tan máu bẩm sinh. Nếu có bất cứ thắc mắc nào cần bác sĩ Phương Hoa tư vấn, vui lòng liên hệ.

Bác sĩ Phương Hoa
Chuyên gia di truyền y học
Liên hệ với bác sĩ





Website này được xây dựng với mong muốn lan tỏa những thông tin hữu ích trong lĩnh vực di truyền học, trở thành kênh kết nối chuyên môn giữa các bác sĩ, chuyên gia y tế nhằm mang lại sự hỗ trợ tốt nhất cho người bệnh.